Contracting Considerations & the Safety of Medical Devices
HealthTrust’s Physician Services team provides a monthly review of FDA Premarket Approvals (PMAs) and 510(k)s for physician preference items. In support of its contracting efforts related to national agreements, these compendiums—along with new technology summaries and clinical evidence reviews (CERs) for implantable medical devices and clinically sensitive products—are made available to members through the secure HealthTrust member portal. Physicians in the relevant specialty who are part of HealthTrust’s Physician Advisors program provide input on the CERs, which are a critical component of HealthTrust’s contracting process.
Have you ever wondered how the process works for getting a device to market? How do healthcare providers know before they buy if a device is worth all of the marketing hype? This type of checks and balances system is the responsibility of the FDA’s Center for Devices and Radiological Health (CDRH), which provides premarket review and approval of all medical devices, as well as oversight of the manufacturing, performance and safety of related devices.
The FDA regulates devices and safeguards customers with one of two types of approval processes—the PMA or 510(k).
• PMAs require manufacturers to submit an application before they market any new products that contain new materials or differ in design from products already on the market. The application requires valid scientific evidence collected from human clinical trials showing the device is safe and effective for its intended use.
> Examples of PMAs include digital mammography, minimally invasive and non-invasive glucose testing devices, implanted defibrillators and implantable middle ear devices.
• 510(k), a section of the Food, Drug & Cosmetic Act, established the risk-based device classification system for medical devices. Before a manufacturer can market a medical device in the U.S., it must demonstrate to the FDA’s satisfaction that the device is substantially equivalent to, or as safe and effective as, a device already on the market. If the FDA rules the device is “substantially equivalent,” the manufacturer can market the device.
> Examples of 510(k)s include X-ray machines, dialysis machines, fetal monitors, lithotripsy machines and muscle stimulators.
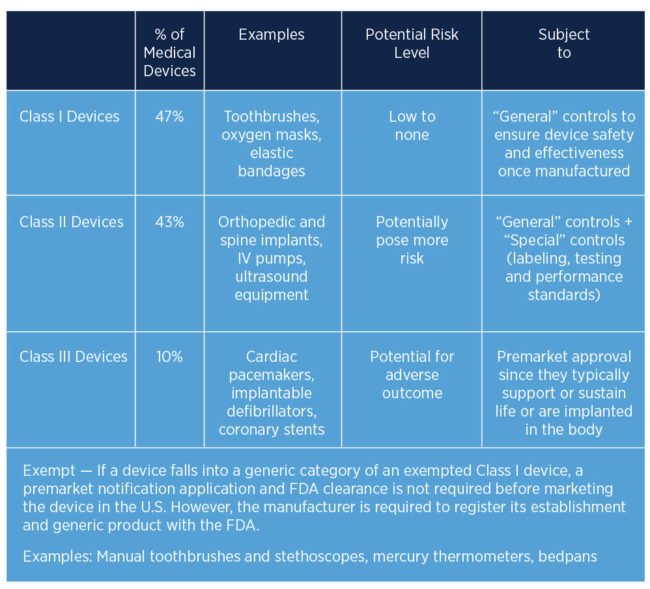
Device Classifications
The classifications of medical devices are based upon the degree of control necessary to ensure the various types of devices are safe and effective before they are marketed to the public. As device class increases from I to III, the regulatory controls also increase, with Class III devices subject to the greatest number of regulations.
Ongoing Protections
The FDA monitors reports of adverse events and recalls related to medical devices. When it perceives the health and safety of patients or product users poses a potentially significant risk, it issues alerts to health professionals and consumers through the medical device safety section of FDA.gov.
HealthTrust also issues important information related to product recalls and/or safety issues through special editions of its contracting newsletter, The Response.
Share Email Q3 2018, Regulations